Research
Our unit explores gene regulation and genetic variation across diverse contexts, leveraging computational methods and the integration of multi-omics data. With a multifaceted and interdisciplinary approach, we address challenges in molecular biology and genomics from multiple perspectives. Our key research areas are:
a) The genetic basis of gene expression and its interplay with organismal complex traits;
b) The evolution of gene regulatory regions and gene expression itself;
c) The genetic basis of behavioral, neuropsychiatric and neuroimaging-derived complex traits.
Other aspects we explored include post-transcriptional regulation mechanisms, gene function inference and the impact of ancestry on complex traits. The unit also collaborates with several experimental laboratories providing bioinformatics and data-mining expertise.
Genetic basis of gene expression
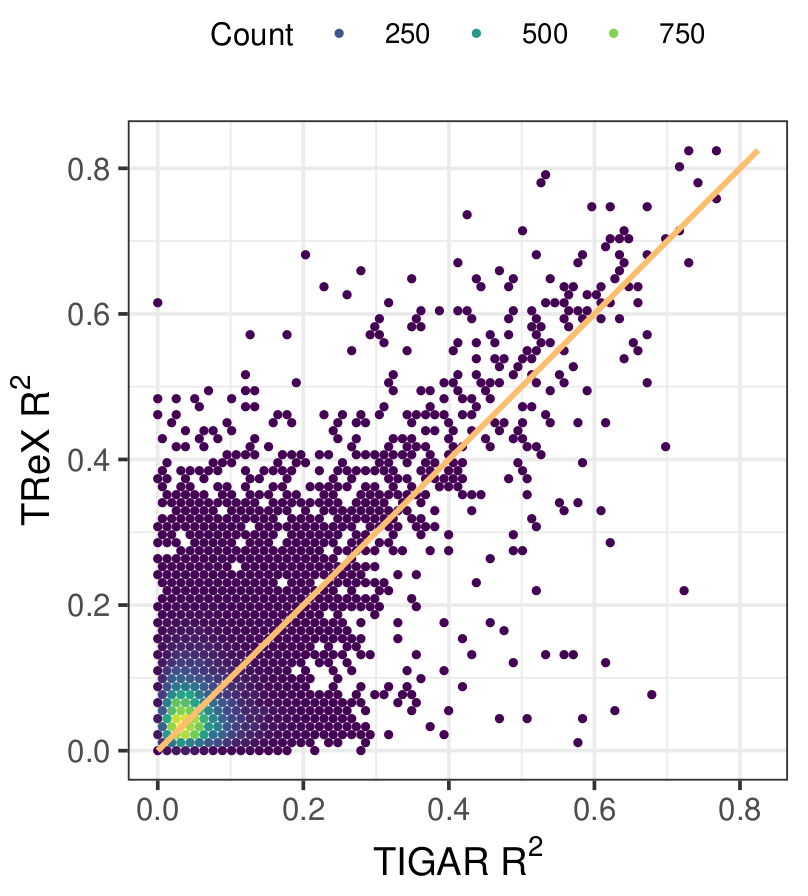
We are interested in how genetic variation shapes the regulation of gene expression and other molecular phenotypes. Our current focus is the prediction of gene expression from sequence and chromatin data in human cell lines. Our approach is based on Total Binding Affinity as a predictor of the binding of transcription factors to cis-regulatory regions. This approach naturally takes into account the contribution of weak binding sites and, from an evolutionary perspective, the phenomenon of binding site turnover.
Relevant publications:
- Marotta F, Mozafari R, Grassi E, Lussana A, Mariella E, Provero P. Prediction of gene expression from regulatory sequence composition enhances transcriptome-wide association studies. Preprint. bioRxiv. 2021. doi: 10.1101/2021.05.11.443571
- Grassi E, Mariella E, Forneris M, Marotta F, Catapano M, Molineris I, Provero P. A functional strategy to characterize expression Quantitative Trait Loci. Hum Genet. 2017 Nov;136(11-12):1477-1487. doi: 10.1007/s00439-017-1849-9
Evolution of gene expression
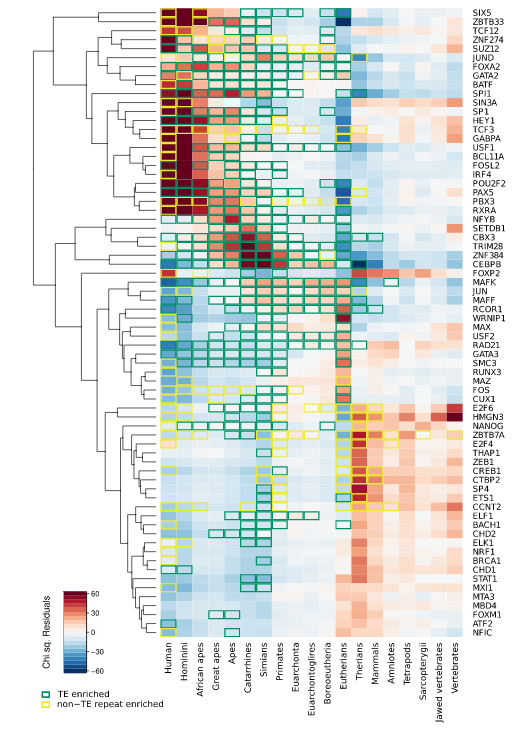
We are interested in the evolution of regulatory regions as a major determinant of anatomical and behavioral differences in humans, great apes and ultimately vertebrates. Our focus is on the role of genome expansion mediated by transposable elements in rewiring the gene regulatory networks through the generation of transcription factor binding sites.
Relevant publications:
- Marnetto D, Mantica F, Molineris I, Grassi E, Pesando I, Provero P. Evolutionary Rewiring of Human Regulatory Networks by Waves of Genome Expansion. Am J Hum Genet. 2018 Feb 1;102(2):207-218. doi: 10.1016/j.ajhg.2017.12.014
- Marnetto D, Molineris I, Grassi E, Provero P. Genome-wide identification and characterization of fixed human-specific regulatory regions. Am J Hum Genet. 2014 Jul 3;95(1):39-48. doi: 10.1016/j.ajhg.2014.05.011
Genetic basis of behavioral, neuropsychiatric and neuroimaging-derived complex traits
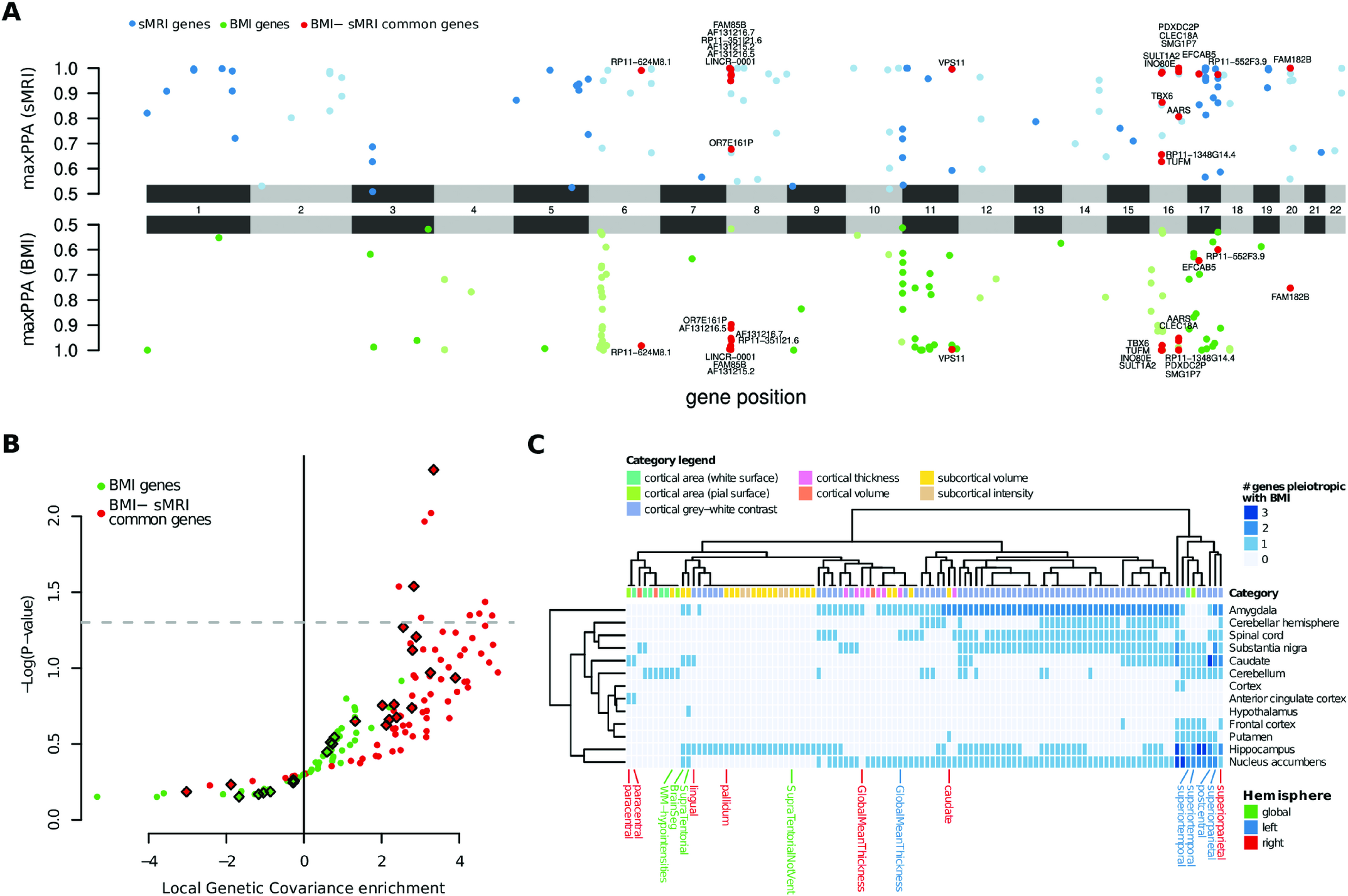
In our most recent publication we integrate molecular biology, neuroimaging, and genomics to understand the molecular underpinnings behind the correlation of brain MRI features and body mass index.
Relevant publications:
- Fusco D, Marinelli C, André M, Troiani L, Noè M, Pizzagalli F, Marnetto D, Provero P. Exploring the molecular basis of the genetic correlation between body mass index and brain morphologicatraits. PLoS Genet. 2025 Apr 10;21(4):e1011658. doi: 10.1371/journal.pgen.1011658
Post-transcriptional regulation
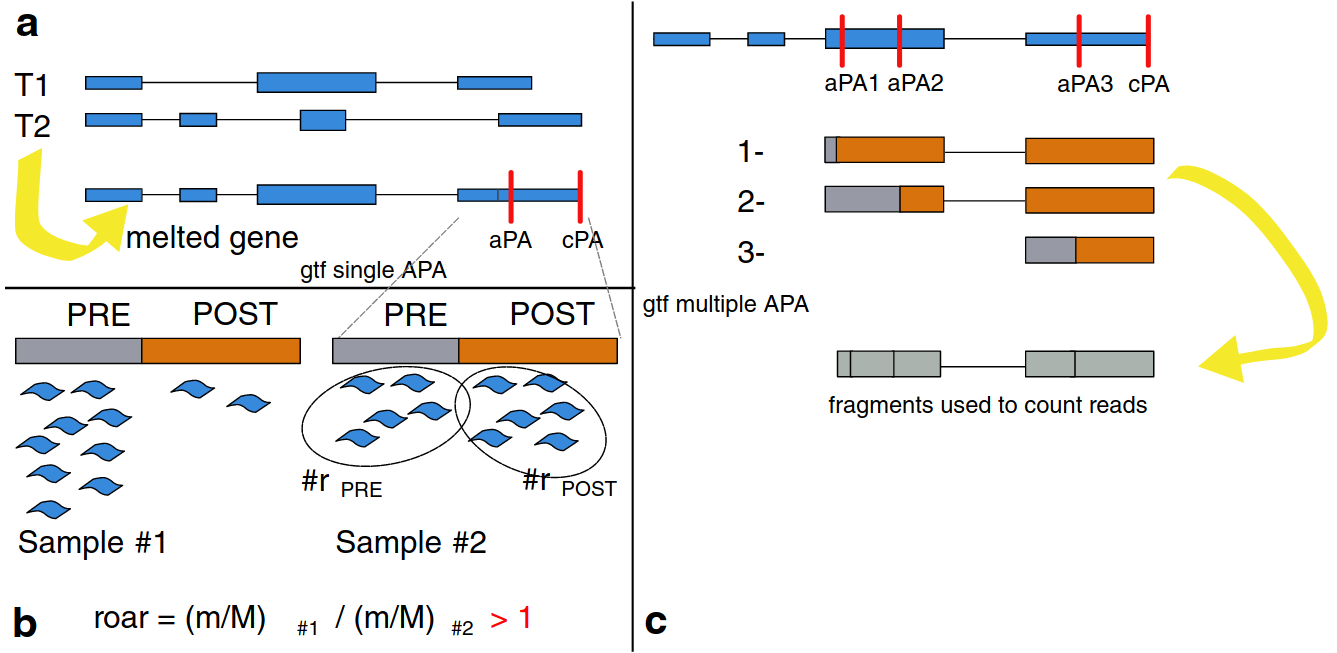 We investigate the role of 3' UTRs in the post-transcriptional regulation of gene expression, in two related aspects: (a) competing endogenous RNAs, where the titration of microRNA abundance by their targets results in a regulatory cross-talk by targets of the same microRNAs that is wholly independent of their gene products (b) shortening of 3' UTR, in which proliferating cells escape regulation by microRNAs and other RNA-binding trans-acting factors by expressing shortened 3' UTR isoforms lacking binding sites for those factors.
We investigate the role of 3' UTRs in the post-transcriptional regulation of gene expression, in two related aspects: (a) competing endogenous RNAs, where the titration of microRNA abundance by their targets results in a regulatory cross-talk by targets of the same microRNAs that is wholly independent of their gene products (b) shortening of 3' UTR, in which proliferating cells escape regulation by microRNAs and other RNA-binding trans-acting factors by expressing shortened 3' UTR isoforms lacking binding sites for those factors.
Relevant publications:
- Mariella E, Marotta F, Grassi E, Gilotto S, Provero P. The Length of the Expressed 3’ UTR Is an Intermediate Molecular Phenotype Linking Genetic Variants to Complex Diseases. Front Genet. 2019 Aug 16;10:714. doi: 10.3389/fgene.2019.00714
- Grassi E, Mariella E, Lembo A, Molineris I, Provero P. Roar: detecting alternative polyadenylation with standard mRNA sequencing libraries. BMC Bioinformatics. 2016 Oct 18;17(1):423. doi: 10.1186/s12859-016-1254-8
Variation across human populations and functional implications
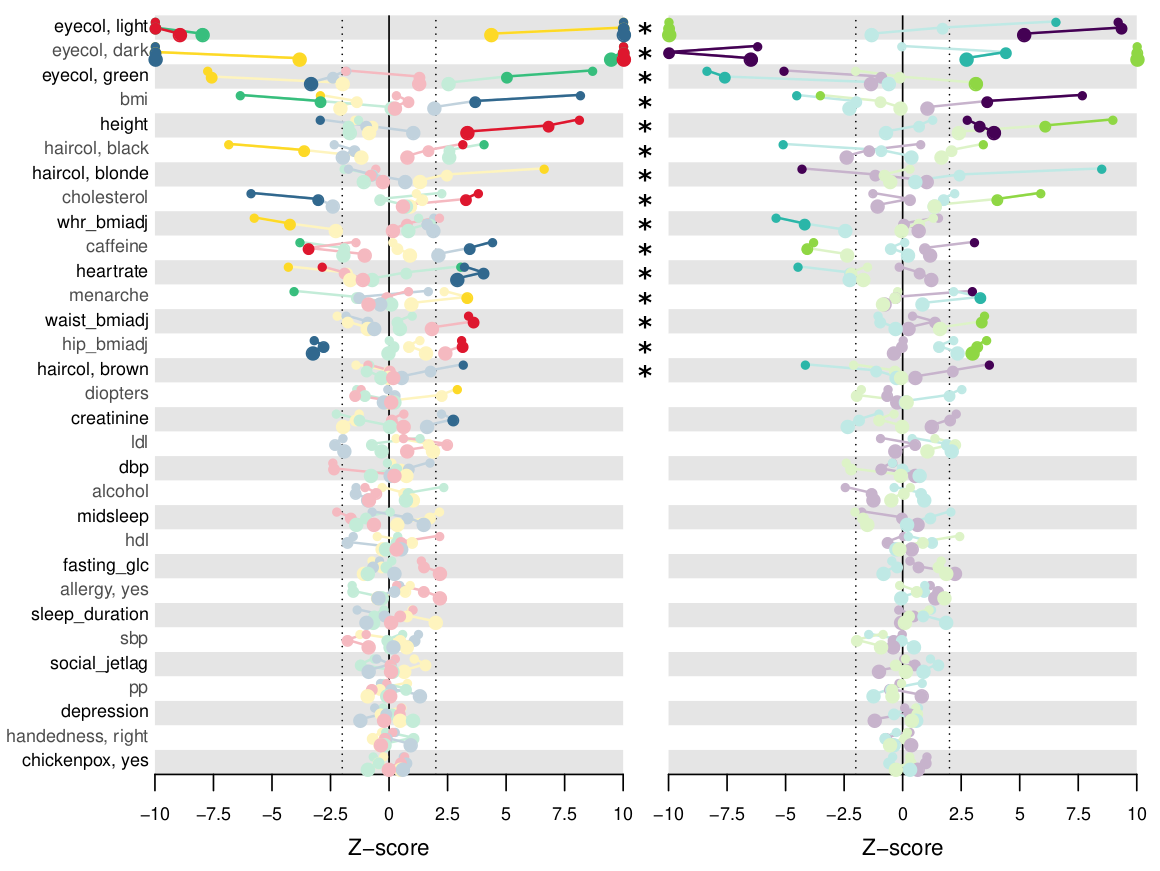 Another interest of our laboratory is the analysis of human variation, for which we developed bioinformatic tools and pipelines. We are currently focused on how genetic variation shapes the complex trait landscape within and across human populations and on the development of approaches to overcome the inherent population-dependency of complex trait predictive methods.
Another interest of our laboratory is the analysis of human variation, for which we developed bioinformatic tools and pipelines. We are currently focused on how genetic variation shapes the complex trait landscape within and across human populations and on the development of approaches to overcome the inherent population-dependency of complex trait predictive methods.
Relevant publications:
- Marnetto D, Pankratov V, Mondal M, Montinaro F, Pärna K, Vallini L, Molinaro L, Saag L, Loog L, Montagnese S, CostaR , Estonian Biobank Research Team, Metspalu M, Eriksson A, Pagani L. Ancestral genomic contributions to complex traits in contemporary Europeans. Curr Biol. 2022 Feb 1:S0960-9822(22)00108-7. doi: 10.1016/j.cub.2022.01.046
- Jagoda E, Marnetto D, Senevirathne G, Gonzalez V, Baid K, Montinaro F, Richard, D, Falzarano D, LeBlanc EV, Colpitts CC, Banerjee A, Pagani, L, Capellini TD. Regulatory dissection of the severe COVID-19 risk locus introgressed by Neanderthals. Elife. 2023 Feb 10;12:e71235. doi: 10.7554/eLife.71235
- Marnetto D, Pärna K, Läll K, Molinaro L, Montinaro F, Haller T, Metspalu M, Mägi R, Fischer K, Pagani L. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat Commun. 2020 Apr 2;11(1):1628. doi: 10.1038/s41467-020-15464-w
- Marnetto D, Huerta-Sánchez E. Haplostrips: revealing population structure through haplotype visualization. Methods Ecol Evol. 2017; 8:1389. doi: 10.1111/2041-210X.12747
Computational methods to infer gene function
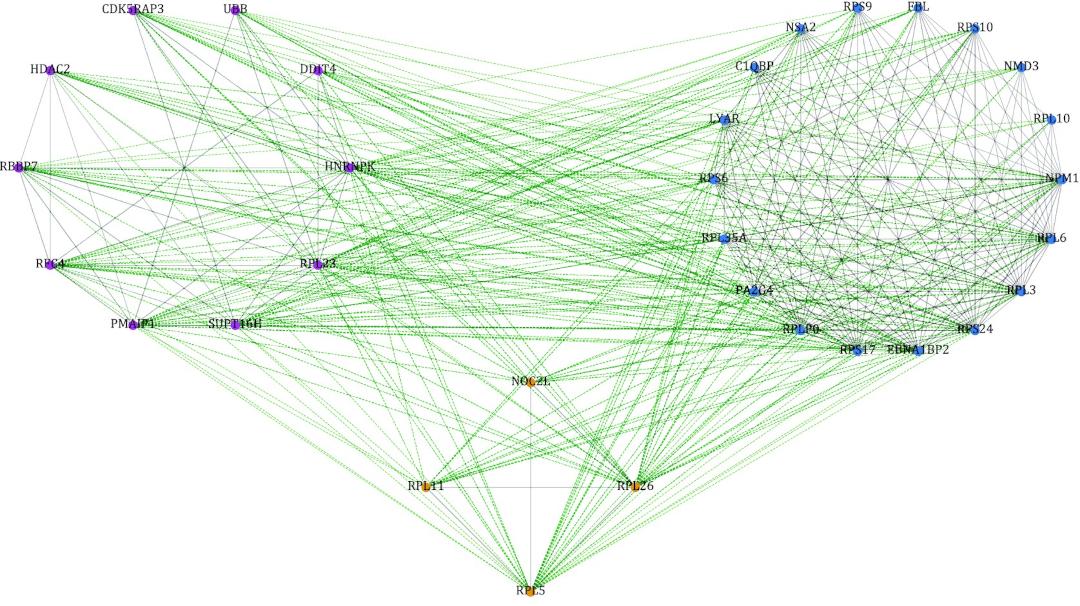 We are interested in computational methods to infer gene function and the phenotypic effects of mutations through the integration of different omics datasets. We develop network analysis methods and machine learning approaches to integrate genomic, transcriptomic, and phenotypic data to understand the role of genetics and gene expression in cancer and diseases.
We are interested in computational methods to infer gene function and the phenotypic effects of mutations through the integration of different omics datasets. We develop network analysis methods and machine learning approaches to integrate genomic, transcriptomic, and phenotypic data to understand the role of genetics and gene expression in cancer and diseases.
Relevant publications:
- Lanciano T, Savino A, Porcu F, Cittaro D, Bonchi F, Provero P. Contrast subgraphs allow comparing homogeneous and heterogeneous networks derived from omics data. Gigascience. 2022;12:giad010. doi: 10.1093/gigascience/giad010
- Draetta EL, Lazarevic D, Provero P, Cittaro D. The frequency of somatic mutations in cancer predicts the phenotypic relevance of germline mutations. Front Genet. 2023 Jan 9;13:1045301. doi: 10.3389/fgene.2022.1045301
- Moiso E, Provero P. Cancer Metabolic Subtypes and Their Association with Molecular and Clinical Features. Cancers (Basel). 2022 Apr 25;14(9):2145. doi: 10.3390/cancers14092145
- Savino A, Provero P, Poli V. Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression. Int J Mol Sci. 2020 Dec 12;21(24):9461. doi: 10.3390/ijms21249461